 ����
����
��������������£������߿ɷ������������
�߹ǹ������δ��ˡ������ѡ�Ѫ�غ�
�����������������ǻ�ڵ�ֱ�������dz�����ԭ��ͷ�����˺���ʶ
�����ߣ�����ѪҺ��θ���������������Դ�Է�����
��ˮ�������������Ҳ���ټ������������Է������Ĵ����߷�����
���Ժ���˥����Խ��Խ��ע�⣻�������Ѫ����Һ���࣬���ۺ��֬��˨�����Լ����˺��Ⱦ��������ɺ��������ۺ�������֪ԭ��
1.�ݿ� ���������ڴ���ʧѪ��ɵĵ�Ѫ��������������������ͣ�ͬʱҲ��ɷ�Ѫ�������١����ڷ�Ѫ�����ļ��ٺ�ԴԴ���ϵؽ�����ѭ����������˨�ӣ��ɶ�����Ѫ�ܴ������谭���彻���Ľ��С��ƻ���Ѫϸ������֯�ֽ���������֧���ܺͷ�СѪ����������ʹëϸѪ��ͨ�����ӣ�����μ��ʳ�Ѫ��ˮ�ף�ʹ���������Ӵ�����ڳ־����ݿ˵Ļ����ϣ������������أ��������Һ����Ѫ�ȣ����ɵ��º��������ۺ�����
2.֬��˨�� ֬��˨���Ƕ���ۺ��IJ���֢�����֬���ο�������С������ʹ֮���š�С֬���ο���ɢ�ںܶ�СѪ�ܣ���ɹ㷺��ѭ��˨����ͬʱ����֬����֬ø�������£��ֽ������֬���ᣬ����ɵĻ�ѧ�����Է�Ӧ���ɵ���
��ˮ���ͷγ�Ѫ���ٴ��ϱ����е���Ѫ֢���Ƿι�����һ����Ҫָ�ꡣ
3.��Һ���� �����ش������У�����Ӧ����Ӧ��ˮ���������ķ�Ӧʱ���Ϊ�־ã�������72h����ˣ��˺������Һ��ʹ����ˮ���������ڣ�������ϸ����Һ����ͬʱ�����������Һ����ϡ��Ѫ�����ף�����Ѫ���Ľ�����ѹ����ʹ
��ˮ�����ء����⣬������౾����ֱ���ܵ����ֲ�ͬԭ�����������ˡ��������ݿ˻�ŧ��֢�ȣ�������������������ˮ�֡���ˣ���ʹ��������Һ������Ҳ�����
��ˮ�������ԣ���Һ�����ڷ������Ժ��������ۺ�������������У���ռ���൱��Ҫ�ĵ�λ���������о�������
��ˮ��ʱ����֫��С���ͷ�ëϸѪ�ܾ�ˮѹ�IJ�𣬷�����֫ëϸѪ��ѹΪ16mmHg��С��ëϸѪ��ѹΪ15.4mmHgʱ���ŷ���ˮ�ף�����ëϸѪ��ѹΪ7.6mmHgʱ��������
��ˮ����
4.��Ⱦ ��ŧ�Ը�Ⱦ��ʹϸ�����ػ�ϸ��������������ѭ�������ڶ��������£������ͷų�Ѫ�ܻ������ʣ���5-�ǰ����鰷�����������Ӱ��ȣ���ʹëϸѪ��ͨ�����ӡ���Ⱦ������ת�����β����Ӷ������ι���˥�ߡ����ݿˡ���Դ��˺ʹ�����Һ�����أ�������ʹ���˷���ŧ��֢���䷢����������˥�ߵĻ��Ƽ�ͼ1��
5.�Դ��� �����Դ��˳�����
��ˮ����������Ϊ�Դ��˿��Լ���ǿ�ҵĽ����嶯������������ĩ��Ѫ���������漴Ѹ�ٷ�����������˥�ߺ�
��ˮ������Ԥ��Ӧ�æ���������������ҩ���ɷ�ֹ������������ִ��˺�
��ˮ���Ļ�Һ�ڵ����ʺ����ܸߣ��ʳ���ѹ��ˮ���⣬��������ͨ��ˮ�����صĴ��ڡ�
6.���� ������Ϊ������������ۺ�����ԭ��֮һ�������ܵ����ӡ���������������θ�������Ƿdz����ص������С��pH����2.5�����Է����Ҳ��������غ��������ѧ�Է��ͷβ���Ⱦ���Ӷ����º���˥�ߡ�
7.���ж� ����˥��ʱ�����ø�Ũ�������ƣ�������ʹ�÷�����ɷ����������ж�����Ҫ��������������ѹ��������ʱ�䣬������ѹ������ʱ�����������Ի���Ŀ������������ж�ʱ��֧���ܵ���ë�˶����ܵ��������ơ�100%������6h�����ɲ�����֢״�ļ���֧�����ס�Sevittͨ������ʬ����������Ϊ��Ĥ�������Է���Ϊ�˷����ж�������������Ҫ�IJ��������ı���ͨ��-��������ʧ��������ѪҺ�����ε�ˮ�ס����š�ͻ�����ά���������ʹ�������������������࣬�γɾ���Ѫ�������ӣ����Dz��������Եĵ���Ѫ֢����������������ɢ�ϰ���������̼�ų����裬��ʱ��ʹ�����Ũ��������������߶�������ѹ��ֻ�ܼ��ضԷεĶ�����ʵ���пɼ����ﳣ��������ȱ��������ͣ����
��������
�������ƣ�������ڱ��������Ļ��ƻ��������������һЩ����ѧ����������Ļ��ƣ���û��һ���ܽ������еķ��������ÿһ���岡�˵ķ���������������ֻ�����������
1.
��ˮ���IJ��� ���ˡ��ݿ˼������²����ص�ʹ��ѭ��ѪҺ�������㣬����ֱ�����ݺ�ëϸѪ�ܡ�ͨ���������ʣ���Ѫ˨��Ѫ�ܻ������ʻ���֢��Ӧ���ʵ��������-ëϸѪ��Ĥ����ʹ��ͨ�����ӣ�Һ���ɴ�ëϸѪ����©�����ݻ�����У�����
��ˮ�������⣬�ݿˡ����˻������²�����ʹ��Ѫ����ע���㣬�Դ�л���ý��ͣ�����������
��Ѫ�ܾ������Ӷ�����ξ���ѹ���ӡ���Һ������Ҳ�������
��ˮ���IJ�����
���˹۲쵽Ѫѭ���е���ϸ����ѪС�弰��֯����ϸ���У������и�����֢���ʣ�����ϸ������ø��ˮ��ø����֬ø�������DZ��ͷŽ����ѭ���У����ʹ����ëϸѪ��Ĥ�����㷺����ʹͨ�����ӣ������ʡ�Ѫϸ����Һ����©��Ѫ���⡣ͬʱѪС��ɷֽ���鰷��Ѫ���ؼ����ģ�ʹѪ����Ƥϸ��������ϸ����϶�ӿ��������ʵ�����������Ҳ��������
��ˮ�����γɡ�
��ʼ��ˮ��Һ�������ڷ�С������Χ�ķμ�����֯���Ժ������࣬����������ϸ֧���ܣ��������������ݣ�����ͨ��/Ѫ����ע����ʧ�����γɵ���Ѫ֢��
2.����
Ѫ˨�γ� Solliday����Ϊ���������أ����п���ʹ���ڶ���Ӱ������ߣ���ʱ�����ƹ�����Ҳ������ҽԴ�Զ���Ӱ����ӡ�����Ӱ������������շ�ѪС���������γ�Ѫ˨���������������࣬���������ε�С�����������ѭ���ϰ���������ѪС�廹���ͷų�Ѫ���غ��鰷������֧���ܾ��Σ�Ӱ��ε�ͨ�����ܡ���
Ѫ˨�γ�����ά����ԭת��Ϊ��ά����ʱ��Ҳ���ͳ�Ѫ�ܻ����ģ����߿ɸ����ؾֲ�Ѫ�ܼ�֧���ܾ��Σ��ζ���ѹ���ߣ�����ëϸѪ��ͨ�����ӣ��Ӷ��������ݼ����ʳ�Ѫ��ˮ�ס���������ά���׳��������⣬˨����Ӱ���Ӫ��Ѫ�ܵ�Ѫ����ע���������֯�ṹ�ƻ������ʹ��˳Ӧ�Խ��ͣ��������������ۺ�������Malik����Ϊ��
Ѫ˨�γ�������Ѫ������Ѫͬʱ���ڣ��Żᷢ�����������ۺ�����
3.���ݱ�������������ɼ��� �����������ۺ�������ʱ������I�ͷ�����Ƥϸ���������ƻ����������ص�����ëϸѪ����Ѫ�����ϵ������ԣ����һ������ɢ�����Ƥϸ���ֻ������ƻ��˵�I����Ƥϸ��������ֱ��Ӱ���˷��ݱ���������ʵ����������������⣬���ڷ���ˮ��Һ���ӯ�����ݱ���������ʵĻ������н��ͣ����ݽ�����ή������˷����������½��������������ѡ�
���ˡ����������������Ժ����ĺ��������ۺ�����������һ��DZ���ڣ�����������ʵİ�˥����18��24h��������ʱ���Ͻ��ƣ���ˣ�������Ϊ���������ۺ����ķ����������ڱ���������ʲ����������¡�
4.ARDS�IJ����������ɶ�����֢ϸ��(����ϸ������������ϸ�����ܰ�ϸ����)�鵼�ķ���ֲ����Է�Ӧ����֢��Ӧʧ�����µķ�ëϸѪ��Ĥ���ˡ�����Ҫ��������Ϊ�ɷ�Ѫ��ͨ�����߶����µķ�������Һ�и��������ʵ�
��ˮ������Ĥ�γɣ��ɰ��зμ�����ά����
�ڳ����ٴ�֢״���ǰ18h�ڣ��δ�����ֲ�����������������ɢ�ڵij�Ѫ��
�β����������ݿ˺�18��72h�������أ�����Ϊ������Ҷ�ʳ�Ѫ�Բ��䡣���������طξ�����Ѫ��ɢ�ڵ�Ѫ˨˨���γɡ�����ˮ�ס�Ѫ�ܺ�֧������Χ��Ѫ�ͷ��ݳ�Ѫ��72h��ɼ���Ĥ��֧���ܷ��ף��̶�������ɢ����ά���Ըı䣬��������ֳ�Ըı��ͬʱ�����ڡ������ı���ص�ɹ������£�
(1)������(24��48h)�����ݺͼ���ˮ�ף�ëϸѪ�ܳ�Ѫ��I�ͷ���ϸ���ƻ���������Ĥ�γɡ�ˮ��Һ�ĵ��������ɷ���Ѫ������ơ���Ѫ�ܵ���Ƥϸ���������������ģ�δ��ϸ�����Ӵ�ȱ�ڡ������ڼ����ڿɷ��ֺ�ϸ������ʾ��Ѫ����Ƥ�������ݵ�©�졣����������Ƥϸ����ǿ����������ʹ��Ƥ����ݵ������Ա�������
(2)ϸ����ֳ��(3��7��)������ϸ������������ϸ������μ������Ĥ����������ϸ���������ں���Ϊ�������Ӧ����ʼѸ����ֳ�������������δ�ܵõ����ƶ����������������Դ̼��йء��ڴ��ڣ�����ϸ�������ڷ�Ѫ����Ƥϸ�����棬��Ѫ��
Ѫ˨�γ�����ʵ�ʵĸı����Ϊ��Ƥ�������������״�Ѫ�ܴ�Ϊ���ٻ��ܼ�ѹ�����ݡ����ڵļ����״�������ˮ���Լ�ϸ����ֳ��
(3)��ά��ֳ��(��7��10��)����Ĥ�ͷ��ݸ�����ά�������ݹ���ά����
(4)�������������ݳ����ţ����ݿ����������ݱ���ëϸѪ�ܸ������������֢��ͷ��ݼ�ëϸѪ����Ƥϸ�����ͣ��ڷ���ëϸѪ���ڿɼ�����ά���ס�ѪС�塢��ϸ���Ͱ�ϸ���������ַ���֯�У���ϸ���������ӣ��ں�ϸ���ͷ��ݱ��棬�ɼ�����ά�������ʸ��š�����ϸ���ĺ�Ⱦɫ�ʱ�֣����ܿ�϶���������������һ���ʧ�����������ţ������ṹ�ƻ������ſ��������Ԣ���ϸ���İ����г��ִ�С���ȵĿ��ݡ��ڷ���ǻ�ڣ��ɼ��������ŵİ�ϸ������ϸ��������ϸ��������Ģ���ϸ�����еķ���ǻ�л�����ˮ��Һ�������Һ��������μ��ʲ�����֯��϶�������ɳ��ֲ�ͬ�̶�ˮ�ף�ż��������ά�ͽ�ԭ��ά����ϡ������ҡ��������ش����ݽṹģ���������壬������Ƥϸ���ı���ɱ��ݺύ������ά�������ǡ�
����
���ƣ�ARDS����Ӧ��������ԭ��������ֹ���������չ�������ȵ���Ҫ��ʱ������������ȱ���������ƹ����в�Ӧ��ARDS�����Դ�����Ӧ������Ϊ�����������ϰ��ۺ���(MODS)��һ����ɲ��֡��ں���֧�������У�Ҫ��ֹ���������·�����(VILI)���������̷���Ⱦ�����ж��Ȳ���֢�ķ��������ݷ����˵ķ������ƣ�̽���µ�ҩ������Ҳ���о�����Ҫ����
1.һ������ ARDS���ߴ��ڸߴ�л״̬��Ӧ��ʱ���������ߵ��ס���֬��Ӫ�����ʡ�Ӧ�������ǿ������Ӫ��֧�֣����ǻ���������Ӧ�����˷��ڰ���λ����
�������㡢ʪ���Ĵ����ڣ���������Կ�����ʹ����ʹ���˿��ԣ�Ӧ��������������̵����֧���ܾ������������п���̵��
2.���ƾ�����Һ�� һ��Ӧ�ʵ�����Һ���������ͷ�Ѫ���ھ�ˮѹ����Һ�����룬�ڱ�֤Ѫ�������ȶ�Ѫѹǰ���£�Ҫ�����Һ����ȸ�ƽ��(-500��-1000ml/d)������Ƥϸ��ͨ������ʱ����������������ڣ����ط�ˮ�ף�����ARDS������Ѫ�嵰��Ũ������ʱ���˸�����Һ��ʹ��С����Шѹ(PAWP)ά����1.37��1.57kPa(14��16cmH2O)��һ����Һ��������1ml/(kg��h)��
3.ҩ������
(1)�������ƣ���������ARDS��ͨ�����ص��ۺ����á���Ƥ�ʼ��ؿ��Խ��ͷ�ëϸѪ��ͨ�ԣ���������������μ���ˮ����Ĥ���γ����µ���ɢ�ϰ���ͬʱ��Ƥ�ʼ��ؿ������ӷ��ݱ���������ʵ����ɣ����ͱ������������ٷ���ή�����µķ��ڷ�����
��Ӧ֤�У�ARDS������ά��ֳ�ڡ�֬��˨�������ARDS�����������ס����������������˺��ж����������롢ŧ�����ݿ˲�����ARDS��
��������ARDS��ԭ�������ڡ�����������Ƴ̣���ν����Ӧ�ڷ���ëϸѪ����Ĥ��������ǰ�����⼤������ARDS�Ļ���֮һ��GC-GCR��������������Ҫ�Ŀ������ӣ�GC-GCR�Զ�������������ؼ���������ã���GC-GCRЧӦֻ��������֢���ӵ��ͷţ������ͷŵ����Խ�������Ϊ�������ڴ�ԭ�����ARDS���������硣
��������ARDS��ע�������ARDS������Ҫ�ۺ����ơ���������ԭ���������ر��ǿ��Ƹ�Ⱦ������ͨ������֯��������ֹ��һ�������˺ͷ�ˮ����Ŀǰ���Ƶ���Ҫԭ����������ARDS�������е�һ�����ڡ���ע��Ԥ������ټ��صIJ���֢�������Ⱦ��ɢ��̷��Ը�Ⱦ����������Ѫ�������������½��ȡ�
(2)��Ѫ��ҩ���Ѫ��ҩ����н��ͷζ���ѹ�����������Ҹ��ɣ����������������ã�������ARDS��Ҫ����߷�Ѫ����ע�����������ͣ�����ȫ�����Ϲ��ܡ�
��Ѫ��ҩ���������ѡ���ԣ��ȿ������Ѫ�����Ž��ͷζ���ѹ��Ҳ������ȫ��Ѫ�����Ž���ȫ��Ѫѹ����
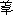
ͪ�����Ѫ�����ţ��ζ���ѹ�½���ȫ��Ѫѹ�½����������������ߣ����Ϲ��ܶ�����������ζ���ѹ�½��������������ߵ��������Ե�ȫ��Ѫѹ�½������Ϲ��ܶ���Ѫ��ҩ��Ե����Է�Ѫ�������������ã����Ƶ�ͨ������Ѫ��������ʹͨ����������ѪҺ���ͨ����������ʹ��ͨ����Ѫ�������ӣ�����������ѪҺ�������½����˼����ڷ������ӡ������ơ���
ͪ�Ⱦ����������÷�����
����һ����������˥�ڼ��̣�ֻ�����ڷ�ѭ�������Ѫ�����ţ�����������ѭ�����ʲ�����Ѫѹ�½������������ҩ��NOֻ����ε�ͨ��������������ͨ������������������ѪҺ���������NO�������Ϲ��ܣ�Ҳ���Ե������ARDS�����ٴ������ʡ�����NO�������ǽ���ARDS�εĵ�һ����Ѫ�����������ã���������ѭ������Ѫѹ�½�������һ����ǡ��NO����ARDS�����ơ������о�֤����ARDS������ԭ����Ҫ�Ƕ����ٹ����ϰ��ۺ���(MODS)������NO��������ѭ��Ѫ�ܸ���ȫ��ѭ��������������θ���������ࡢ����ȹ��ܲ�����������������ȱѪ�ٽ�ϸ����λ���⽫������ʹ�Ѿ����Ƶķι������±仵��
(3) �����ɻ���������������������������绯ø(SOD)��
��������ø(CAT)���ɷ�ֹO
2��H
2O
2��������������ļ��Է����ˣ����������O
2��OH�IJ�����PMN����������ά����E����һ����������Ч�ܡ�֬����ø�ͻ�����ø;�����Ƽ����粼��ҵȿ�ʹѪ˨��A
2��ǰ�����ؼ��٣����Ʋ�����PMN��ϣ���ֹPMN�ڷ��ھۼ���
(4)�������ƣ���ͨ���к��²����ӣ��Կ����Խ��ʺ�����ЧӦϸ�������� ARDS��Ŀǰ�о��϶���п��ڶ��ؿ��壬��TNF��IL-1��IL-6��IL-8���Լ���ϸ��𤸽���ӵĿ����ҩ����ڲ���ALI�Ľ���ʮ���ڶ࣬����֮��Ĺ�ϵ��Ӱ������ʮ�ָ��ӣ����Խ��������ijһ���ʺ����ؽ��и�Ԥ����ЧӦʮ�����ޡ�
4.��еͨ�� һ��ȷ���Ҫ���Ǽ��ͼ�ǿ���Ʋ��������������п�����������̵Һ������������������С������Чǻ�ͺ�����������������ʱӦ�û�е����ͨ���������еͨ������ARDS���ڵ������о����֣�ARDSʱ�������˵ķֲ������Ǿ��ȵģ�������������ݱ��ݣ�����������ݱ��ֿ��ź�����ͨ����ͨ��������Ӱ�����·������ڹ㷺�ķ�ˮ�ͷβ��ţ������Ϸ�������ͨ���Ϻõķ��ݡ���CTɨ��֤ʵ�˲�ͬ��λ�´������������Է�Һ���������ARDSʱ�������彻���ķ�������������������35%��50%������ARDS��������20%����ʹ��������ȫ��ͨ���ij��泱����ʱ���ᵼ�»�еͨ������Է�����VALI��VALI�ķ�����Ϊ4%��15%������Ϊ���ٷ��������壺�����μ��ʡ��ݸ���Ƥ���������ء��������İ���Ĥ������Լ�ȫ��������˨��(��ξ������ԡ���״����˨����)���������Է�ʵ�����ˣ�����������Ƥ��Ѫ����Ƥ���ˡ�ˮ�ס���Ѫ������Ĥ�γɡ���ϸ�������ݲ��ŵȡ������������ױ����������ڸǣ������Է�ʵ�������뼱�Է�����/���Ժ��������ۺ���(ALI/ARDS)�ȵIJ����ı����ƣ���ʱ�������֡�����������ʶ����Ҫ�������ĸ�����ѹ��������������Ļ�еͨ�����Լ��������������ܹ����Ʒ��ݿ��ѹ(Ϊ����ĩ����ѹ����ǻ��ѹ֮��)�;������ٺ��������з�����ѹ�����ݻ��仯�ķ��������Զ���Ѫ��Ϊ��ת��ͨ��ѹ�����ƵIJ��ԡ�
(1)ARDS�Ļ�еͨ�����ԣ�Ŀǰ�ṩ�Ļ�еͨ�����ԣ���Ҫ�����¼������棺��Ϊ��С���ݿ��ѹ��������ݹ������ţ��ı��������ݻ�Ŀ����Ϊѹ��Ŀ���͡��ٴ���������ƽ̨ѹΪָ�꣬ʹ�����2.94��3.43kPa(30��35cmH
2O)����Ϊ������ݹ������ţ��ɽ���ͨ���������������Ը�̼��Ѫ֢(permissive hypercapnia)���ԡ��ۿ�ͨ���ı����ʱ�ȣ����÷���ͨ��(IRV)���������Ʒ���ͨ��(VC-IRV)��ѹ�����Ʒ���ͨ��(PC-IRV)�ķ�������������ѹ(PIP)���������ƽ��ѹ(Paw)�γ��ʵ�ˮƽ����Դ��PEEP(PEEPi)������������ή�ݷ��ݸ��ţ����ٷ��ݱ���������ʶ�ʧ���ܾ������ٻ�еͨ����ǿ���ԣ���ǿ�������������ã��ٽ���еͨ��������������Э�������Ƶ��ͨ��(HFOV)��ѹ���ͷ�ͨ��(APRV)�ȼ�����Ӧ�á���Ӧ�÷���ѧ����ȷ����PEEPˮƽ��Ѱ�ҡ����PEEP����ʹ֮�ȿ��Է�ֹ����ĩ����ή�ݣ���ͬʱ����������ӷ���ѹ������ARDS�ķ�����״̬���没�̱仯��ǿ����̬������⣬���Լ�ʱ����ͨ������������һЩ����֧�ּ��������������ڴ�������(TGI)������λͨ��(prone positioning)��Һ��ͨ��(LV)���������彻������(����Ĥ���ϡ���ECMO������ȥ��
������̼����ECCO
2-R��Ѫ�������ϼ�������IVOX)�ȣ������о�����ʾ��һ�����ٴ�Ӧ��ǰ����
ARDS��ͳ�����ͨ��Gattiononi�ȸ��ݷ���֯����̶ȵIJ�ͬ��ARDS���ߵķ���֯��Ϊ3���֡��ٷ���֯����ʵ�䣺������ͨ�����ܣ��ڷ���֯�����������Ͷ����·δ��壻�۷���֯�Ľṹ������������������ͨ��/Ѫ����ֵ��Ȼ�����о����֣� ARDS���߷εIJ����ı��������Եģ�������֮��ķ���֯˳Ӧ�Դ����Žϴ�IJ��죬���ڷβ��ż�ˮ�ף�ʹ�ε���Ч�����ݻ����Լ��٣������߽�Ϊ�������ݻ���25%����ˣ�������������ͨ���Ʊص�������ѹ���������ݹ�������������ݻ��ˡ��ͳ�����ͨ���Ǽ��ٷ��ݹ���������ķ���֮һ��Lee�ȶ���������Ҫʹ�ú������Ļ��߷ֱ�ʹ�õͳ�����(6ml/kg)�ͱ�������(12ml/kg)���л�еͨ����������֣��ͳ�����ͨ���黼�ߵķβ���Ⱦ�ķ����ʵͼ����ܲ�ܵ�ʱ���������̣����߲����ʽ��͡�Ȼ�������Զ����ĵ��о������ʾ����ѹ�ͳ�����ͨ��[��������8ml/kg��������ѹ��2.94kPa(30cmH2O)]�볣���еͨ��[������Ϊ10��15ml/kg��������ѹ��4.9kPa(50cmH2O)]��ȣ����ߵIJ����ʼ�����֢�������ԵIJ�𡣴��⣬�ͳ�����ͨ��������С�����ıպϡ������Էβ����Լ��������������CO2Ѫ֢�ȡ���ͳ��ΪӦ����������ͨ��Ƶ����ά��������PaCO2ˮƽ����ͨ��Ƶ������(��25��30��/min)��ʹ����ʱ�����̣����·�������������������Դ��PEEP�����������ѭ��ϵͳ��ЧӦ����Ƶ�ʹ��죬��ʹ����Դ��PEEP��Ҳ��Ӱ������ܡ���ˣ��ͳ�����ͨ����ARDS�����л�Ӧ���������α�����ͨ�����ԣ����ܻ�ﵽȡ�����̵����á�
ARDS�������Ը�CO2Ѫ֢�����Ը�̼��Ѫ֢������С���������ͷ���ͨ������������һ���̶ȵĸ�̼��Ѫ֢(һ��PaCO2���˸���10.7��13.3kPa(80��100mmHg)��pHֵ���˵���7.20����pHֵ���Ϳɲ��Լ���������ۻ����ǣ��ٶԻ�еͨ�������ݻ��Է����˵����ӣ���ͼ��������ʱ���ݵĹ������ţ�����ΪѪ��һ���̶ȵĸ�̼��Ѫ֢�͵�pHֵ�������������������ˣ������ԡ�����������״��������һ��Ȩ������֮�����Ѷ�Ϊ֮������ѡ����ʹ����Ҫ�����丱���ã��ر�ע���ų�����֤(�ڸ�ѹ�������Ĺ��ܲ�ȫ��)�������Ը�CO2Ѫ֢�ǵͳ�����ͨ�����ɱ���Ľ��������������£���CO2Ѫ֢�Ի����Σ����Ҫͨ������������Ļ��ƣ��ٽ�����Һ��pHֵ�������������ʱ��ͨ������PaO2���������صĵ���Ѫ֢������CO2Ѫ֢�����Ի����Ӱ�첢�����ء�
��еͨ������������״̬������ͬ���ٻ�еͨ��ʱ��ͨ�������Լ���CO2Ѫ֢���µĵ���Ѫ֢��ͨ�����ƽ��о������ڵͳ�����ͨ���������ĸ�CO2Ѫ֢������ζ�Ż��ߺ���˥�ߵļ��أ���һ���̶ȵĸ�CO2Ѫ֢�Ի��岢��̫������ܶ��Ѿ����������˵Ļ��ߣ���ʹ�û�еͨ��ʹCO2�ָ���������������ԭ�еķ����ˡ�Ϊ�ˣ���������������Ը�CO2Ѫ֢�ĸ��������PaCO2���ߵļ����Լ��������ٶȻ��������顣һ����Ϊ�����߿�������10��12h�������ߵ�CO2Ѫ֢������13.3kPa(100mmHg)Ҳδ���������Ե�Ѫ������ѧ�ĸı䡣Carvalho�ȶ�25�����߿����յ���CO2Ѫ֢������PaCO2�������ߵij��ڿ��������ص�Ѫ������ѧ�仯����36��48h��Ѫ������ѧ�ָ��������������������ֻҪ���ഢ�����ܼ�Ѫ������������������CO2��ɱ����ܡ�
�����������ʱ�������Ը�CO2Ѫ֢Ӧ���ã���ȱѪ�����ಡ����������Ĺ���˥�ߣ��۷ζ�����ѹ���������ˡ�
(2)ARDS���л�еͨ��ʱ������(VT)��ѡ��Ŀǰ�Ƽ�С������ͨ��(VT 6��8ml/kg)��VT�ĵ����ڶ��ݷ�ʽ��Ӧ�ο�����ƽ̨ѹ(Pplat)��ʹPplat����2.94��3.43kPa(30��35cmH2O)��VT�Ĵ�С�������PEEPˮƽ��������PEEPˮƽ��VT��С����СVTͨ�������£����ʵ����Ӻ���Ƶ����������֤����ͨ������������Ƶ�ʲ��˸���30��/min�����������·����ˡ���ʱ�ɽ��ܵ�ͨ��״̬����ȡ�����Ը�̼��Ѫ֢���ԣ�����һ���̶ȵĸ�̼��Ѫ֢��PaCO2һ�㲻�˸���10.7��13.3kPa(80��100mmHg)��pHֵ���˵���7.20����pH��7.20�ɲ��
(3)ARDS��PEEPˮƽ�ĵ��ڣ�PEEPˮƽ����ԭ���Ǽ���ʹ��˳Ӧ�������ݿ��ţ�ͬʱ�ֲ���ʹ����˳Ӧ�����ݹ������š�һ��ʹ�����ں���ĩѹ��������0.49��1.47kPa(5��15cmH2O)��ȷ�����PEEP�ķ����У����������㲻ͬPEEP�¾�̬˳Ӧ��(Cstat)��(Cst������VT/����ĩ������ѹ������ĩ������ѹ������VT/ƽ̨ѹ����PEEP)��Ѱ����Cstat����������ת�۵����Ӧ��PEEPˮƽ��������ͬ���������¸ı�PEEPˮƽ���۲�������ѹ��ͬ���仯��Ѱ�ҵ���ѹ���ӷ��ȿ�ʼ����PEEP���ӷ��ȵ�ת�۵��Ӧ��PEEPˮƽ��������PEEP��ѡ��Ӧ���ݷξ�̬˳Ӧ�ԡ���̬ѹ��-��������(PV����)�Ĺյ�(inflexion)��������ARDS���ߣ������߳�S�Σ��м䲿�ֽ϶�������ֱ�ߣ����ڶ������ײ���ƽ̹�����м�ֱ�����Ӵ��ֱ��Ϊ�ϡ��¹յ㡣�¹յ���С���������ͱպϵ�ת�۵㣬��PEEP��ֵ�������Ը����¹յ��ֵ���Ա���С�����������Կ��գ���������������ˡ�ͬʱ�������ķ�ѹ��Ҳ��Ӧ�����Ϲյ㣬ʹ��еͨ��ʼ�մ��ڷξ�̬˳Ӧ�Ե�ֱ�߲��֡���Ӧ��ע����ǣ�PEEP�����ЧӦ��Ҫ20��30min�������ʱ�䷽�ɳ�ֵر��ֳ�����
Ȼ����Ӧ��PV����ѡ��PEEP����һ���ľ����ԣ���ARDS���ߵ�PV���ߵĹյ������Ų���IJ�ͬ�ζ����ϱ仯��չ�ģ���Щ���������ⲻ���յ㡣�ڻ���PV������Ҫ������������Σ�ػ��߿�����������Σ�ա���ˣ����о������Ӧ�ö�̬ѹ���ݻ���(PVloop)��ȷ�����PEEP��ʹ�÷��㣬��������������PVloop����������������PV������̬���������Ƶ��ϡ��¹յ㡣���ڲⶨPVloopʱӦע������������٣���������ͨ�����Լ�������������ɷֵ�Ӱ�죬����������������Ļ��������������ܡ�
���⣬PEEP��ѡ��Ӧ���Ƕ�����ܵ�Ӱ�죬PEEP����Ӱ�������ÿһ��������Ӱ�������ǰ�����Լ�������������ܣ�����Ӱ������Ѫ������ˣ���Ӧ��PEEPʱӦ��ǿ�Ի�������ܵļ�⣻ͬʱ�����������������DZز����ٵġ�������PEEP��Ӧ�ÿ���ɶԻ������������ж��ϵ����ѣ�һ����ΪPEEP��0.98kPa(10cmH2O)��Ѫ������ѧ��������Ӱ�졣���ڽ���Ѫ������ѧ�ⶨʱӦע��ξֲ�˳Ӧ�Ըı䡢�ζ������ܼ�˵�λ�á�ƽ������ѹ�ߵ͡���Դ��PEEP�����ض�ѹ���ⶨ��Ӱ�졣��ˣ����������ɣ����ڳ�����ϵ�����£���ʱ��������������ѻ���Ѫѹ�������ߣ�����PEEP��Ѫ������ѧ��Ӱ�죻���ѻ���������Ѫѹ���ߣ�����ʾ��Դ��PEEP�Ĵ��ڣ����������зζ������ܣ��ɸ��貹Һ���飬�۲���Ѫ�ܷ�Ӧ�����߳���Ҳ����������״̬��ȫ����Ч�ķ����������������������ͬ�������������������������ߵİڶ�����ʾ��Ѫ�������㡣
(4)ѹ��Ԥ��ͨ���뷴��ͨ������ARDS��ѹ��Ԥ��ͨ���Ѿ����ٴ�����ѡ��ͨ��ģʽ��ѹ��Ԥ��ͨ��������ѹ������ͨ����ѹ�����Ʒ���ͨ��������ѹ���ͷ�ͨ����ѹ�����Ƽ�϶ָ��ͨ����ѹ��֧��ͨ���ȡ�ѹ��Ԥ��ͨ�����ŵ��ǻ�еͨ���뻼�ߵ�ͬ���Ժã������˷��ݻ��˵ķ����ʡ������ϣ�����ͨ��IRVͨ������I��E�����������������٣��Ӷ�������PIP������ƽ������ѹ�������ڽϵ͵�PEEPˮƽ�Ͻ��ͷ��ڷ�����ͬʱʹή�ݵķ����������ţ��������彻����IRVͨ����������ʱ�䣬�ȶ��ε�λ���������彻������������������ֲ����ڷ��ݹر�ʱ�ݻ���С���Ӷ����·α����������ȱ��������ȶ��Ը��ţ�����ΪPEEP���滻������
����ARDS������Ӧ�÷���ͨ�����������٣���ȻAbel�ȷ��ַ���ͨ�����Խ���ARDS���ߵIJ����ʣ��������ų����������ƴ�ʩ�ĸ���������������Ч�����ҿ��ܳ�����Դ��PEEP�������Ⱥ�����ʱ�����������ء���Ѫ�����ƵȲ���֢�����ֻ��߿�����Ҫʹ��������ʩ�з���ͨ���������о������֣�ƽ������ѹ�����Dz����ݻ��˵���Ҫԭ���ɴ������˷���ͨ�����ٴ�Ӧ�á�
(5)����λͨ���������Է�Ѫ��������һ�������������ƣ�������ͨ���Ϻõķ���֯��Ѫ������ˣ���λ�ĸı��ARDS���߷��ڵ�Ѫ���ֲ��������Ե�Ӱ�졣��������λͨ��ʱ����ǰ�����������ǻѹ���ݶȣ���ֵ���ͣ�������֯ˮ���������������ԭ��ʹ�����ķ������ڱպ����ݣ�����ͨ�����㡣������λʱ�����ڸ���λ����֯�ľ���ѹ������һ�£����γɸ�Ϊ���ȵ�ͨ��Ѫ����ֵ���Ӷ��������ϡ�Gattinoni���о�����������λ10min������ʹ����֯���ܶȷ����طֲ���10��45min�����ϸ��ƣ�����λ�䶯ʱ������������Ѫ�����������ٷֲ������ٴ����Ҳ����������λʱ���ڷ������½�����˳Ӧ�����ӣ�����ָ��(PaO2/FiO2)���ӡ�����λ��Ȼ����֢���٣���Ѫ������ѧҲ������Ӱ�졣�����н�Ĥ�����沿ˮ���Լ����ܵ��������ѳ��ı��������ڸ���λ��Ӱ�쾲�����������ڸ�ѹ��Ӧ���á����⣬���ѵĻ���Ӧ�ø���λͨ��ʱ��ʱ��ҪӦ������������ϼ���
(6)ARDS������ѹ���ͷ�ͨ��������ѹ���ͷ�ͨ��(airway pressure release ventilation��APRV) ���������������Ƚ�ǿ�IJ��ˣ������ٽ���еͨ��������������Э�������ٻ�еͨ����ǿ���ԡ�APRV��ԭ�����ں�����·�����÷�ֵ��������ѹ���ͷŻ�ꡣѹ���ͷŻ�꿪��ʱ������Ӵ˻����������������������ʹ����ѹ��FRC���ͣ�PEEP�ͷš�ѹ���ͷŻ�꿪����Ƶ�ʡ�ѹ���ͷ�ʱ��ij��̺��ͷų̶ȶ��ǿɵ��ġ�����ѹ���ͷŵļ�Ъ�ڣ���������ͨ����ֵ������������PEEP��PEEPˮƽҲ�ǿɵ��ġ�APRV�������Ӽ��Ժ���˥���ߵķ���ͨ�����������ϡ�����Ϊ��ѹ���ͷŻ��ر�ʱ��PEEPʹFRC���ӣ�ή�ݷ��ݸ��ţ�������
�������ëϸѪ������ɢ����ѹ���ͷŻ�꿪��ʱ��������PEEPˮƽ���ͣ������������ӣ�CO
2�ų����ࡣ��ѹ���ͷŻ�����¹رպ�PEEPˮƽ��FRC��Ѹ�ٻָ���ԭ��ˮƽ�����Է���ͨ����������ȡ�����ͷ�������APRV��Ƶ�ʣ����ͷ��������ɷ�˳Ӧ�ԡ���������PEEPˮƽ��ѹ���ͷ�ʱ����ͷ��ݶȵȾ����ġ�
(7)ARDS����ͨ������ģʽ��
�������ڴ���(TGI)�������ܲ��������ͨ���ܵ�����˾�¡ͻ1cm����6L/min�������ɼ�����Чǻͨ��(VD)���ٽ�CO2�ų�������TGI��������ʱ��ɷ�Ϊ���࣬������TGI(CTGI)�ͺ�����TGI(expiratory washout��EWO)��ǰ���������ͺ���������������߽��ں�����������������ʱTGI������������Ϊ������������һ���֣�����������������ʹ���ܼ������Ľ�����Чǻ��·��������Χ�����������ٽ�CO2�ų���CTGI�����У�Ϊ����ѧ�������á�������ʱTG1�����������ɳ�ϴ���ܻ�������Чǻ�ڵ�CO2�����ӷ���ͨ������
��Ӧ��ijЩͨ����ʽ(��ѹ������ͨ��)����CTGIʱ��������ѹ�����ᳬ��Ԥ��ѹ��ˮƽ��������ѹ�˵�Σ�գ�����Ѫ������ѧ��������Ӱ�졣CTGI��PCV��ϣ�����Ч�ظ��Ʒ���ͨ����������CO2���ų�����Ѫ������ѧ��������ѧ������Ӱ�죬����Ϊʵʩ�α�������ʱ��ֹCO2���ߵĸ���ͨ���ֶΣ�Ϊ�˷�ֹ����ѹ���ߣ�����ƺ�ʵʩCTGIʱ����PRV��ΪCTGI�ر��IJ��ּ��Կ����DZز����ٵģ�PRV��Ӧ��Ϊ�Ӵ�TGI���٣��Դﵽ����ϴЧ�����ڷ���ͨ��ʱʹ��CTGI�ṩ�˰�ȫ���ϡ�
�ڸ�Ƶͨ��(HFV)��������Krishnan��������˸�Ƶͨ��(HFV)Ӧ���ڼ��Է�����(ALI)��ARDS���ߣ����Ƿ���HFV����������ŵ�:A.ʹ�õͳ��������ڲ�����ι������͵����������ӷ��ں���ĩ�ݻ���B.���Ʒε�ͨ�����������ܣ�ά�ֻ���PaCO2ˮƽ����������ӽ�����ˮƽ��
��Һ��ͨ��(LV)�����Ƚ������������и߶ȿ����ԡ����������ͣ��Ҷ���֯���κ���Һ�塪��ȫ��������(perfluorocty bromide)������ע��Σ�Ȼ�������ѹͨ�����˷�����������ͨ��ʱ������ȡ(PaO2���4��)��CO2���ų��������ͷ��ݱ������������ӷ�˳Ӧ�ԡ��Ӷ��ȿɱ�֤���彻���ֿɿ�������ѹ��VT�ڰ�ȫˮƽ���Ҷ�Ѫ������ѧ��������������Ӱ�졣��ע��Һ�����ڷ���������ΪȫLV����ע��Һ�����ڹ��ܲ���������Ϊ����LV����ȫLV���������ߣ����곫�ò���LV��Һ����Ȼ�ӷ�ԼÿСʱ2ml/kg��LVʱӦ��ʱ����PFCҺ���������ת��ͣ��LV��ֻҪֹͣ����PFCҺ�����ŷ���PFCҺ�Ļӷ�������LV������Զ�תΪ����ͨ����
�������������彻����ѡ��ECMO��ECCO2-R��IVOXS���з�ʽ�������еͨ�����·����ˣ�ʹ�γ����Ϣ���Է������彻��װ���ṩ���߱�Ҫ�����Ϻ�CO2�ų�����Ŀǰ��δ�ռ�Ӧ�ã�ȱ�����飬�д�������о�֤ʵ���ٴ�Ӧ�ü�ֵ��



